






.png)


















Newborn Screening of Inherited Metabolic Diseases by NGS
"Badyhealth" newborn screening of inherited metabolic diseases by NGS, using Uni-medica "All-in-One" mutiplex PCR amplicon library preparation technology and NGS technology, to detect 120+ inherited diseases.


Shenzhen Uni-medica Technology Co., Ltd (Uni-medica) is a national high-tech enterprise in the field of genetic diseases and reproductive health, providing specialized genetic testing for newborns and children. Uni-medica has the international leading strength in high-throughput sequencing technology and bioinformatics data analysis, and is committed to becoming a leader in the field of precise prevention of birth defectsin.
The company's technical research team is located at Harvard University. It has established cooperation with the Hong Kong University of Science. The company owns a leading genetic testing technology, automated bioinformatic analysis system, and leading domestic phenotype and genotype association database,
which can provide automated and localized solutions for genetic disease NGS detection and analysis. It has undertaken scientific research projects such as the Shenzhen Peacock Plan technology research and development project and the Shenzhen core technology breakthrough project, and has applied for more than domestic, international patents and software copyright.
Uni-medica has established a high-quality comprehensive team, including talents from Shenzhen Peacock Program, Doctor of Biomedicine, Genetic interpretation expert. Chief scientists and Academic leaders are from Hong Kong University of Science and Technology and Harvard University, and have brought together outstanding medical experts at home and abroad, and have won high recognition from clinical clients.
Inherited metabolic disease

Amino acid metabolism disease
0
Organic Acid Metabolism
0
Fatty Acid Beta Oxidative Metabolism
0
Lysosomal Storage Disease
0
Carbohydrate Metabolism
0
Treatable metabolic epilepsy
0
Other Genetic disease
0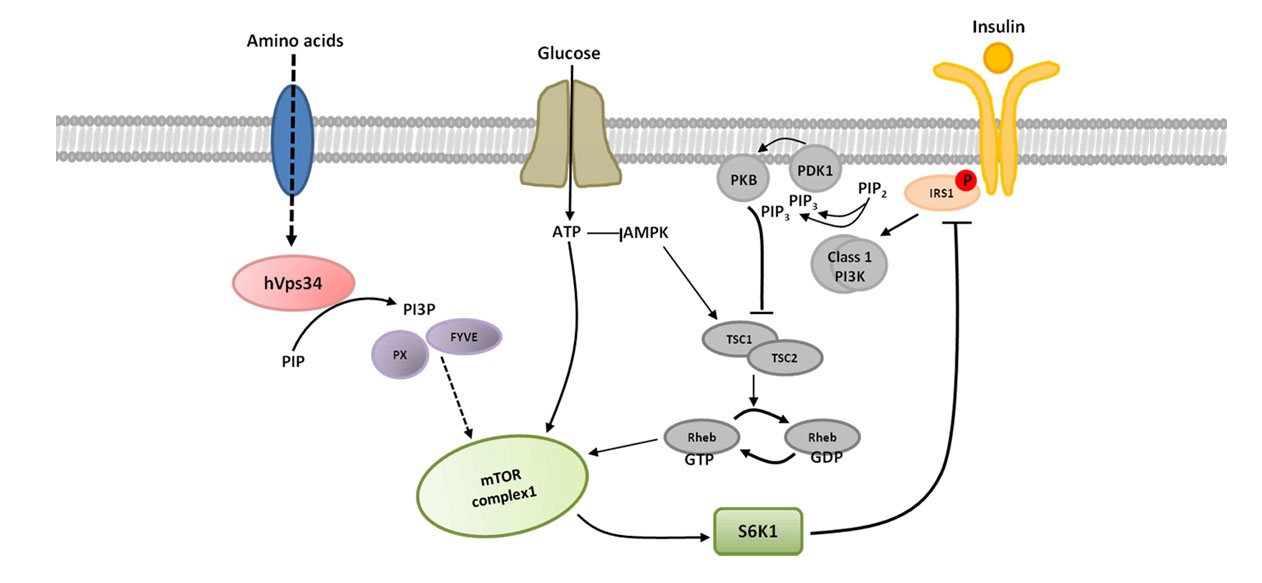
Phenylketonuria
Hyperphenylalaninemia, BH4-deficient, A
Hyperphenylalaninemia, BH4-deficient, C
Hyperphenylalaninemia, mild, non-BH4-deficient
Hyperphenylalaninemia, BH4-deficient, D
Hyperphenylalaninemia, BH4-deficient, B
Dystonia, dopa-responsive, due to sepiapterin reductase deficiency
Tyrosinemia, type I
Tyrosinemia, type II
Tyrosinemia, type III
Maple syrup urine disease, type Ia
Maple syrup urine disease, type Ib
Maple syrup urine disease, type II
Dihydrolipoamide dehydrogenase deficiency
Carbamoylphosphate synthetase I deficiency
Ornithine transcarbamylase deficiency
N-acetylglutamate synthase deficiency
Citrullinemia, type I
Citrullinemia, type II
Argininosuccinic aciduria
Argininemia
Gyrate atrophy of choroid and retina with or without ornithinemia
Hyperornithinemia-hyperammonemia-homocitrullinemia syndrome
Homocystinuria, B6-responsive and nonresponsive types
Homocystinuria due to MTHFR deficiency
Homocystinuria-megaloblastic anemia, cblG complementation type
Homocystinuria-megaloblastic anemia, cblE type
Hypermethioninemia, persistent, autosomal dominant, due to methionine adenosyltransferase I/III deficiency
Glycine N-methyltransferase deficiency
Hypermethioninemia with deficiency of S-adenosylhomocysteine hydrolase
Hyperprolinemia, type I
Glycine encephalopathy

Methylmalonic aciduria and homocystinuria, cblC type
Methylmalonic aciduria and homocystinuria, cblD type
Methylmalonic aciduria and homocystinuria, cblF type
Mental retardation, X-linked 3
Methylmalonic aciduria and homocystinuria, cblJ type
Methylmalonic aciduria, mut(0) type
Methylmalonic aciduria, cblA type
Methylmalonic aciduria, cblB type
Mitochondrial DNA depletion syndrome 5
Mitochondrial DNA depletion syndrome 9
Methylmalonyl-CoA epimerase deficiency
Combined malonic and methylmalonic aciduria
Methylmalonic aciduria, transient, due to transcobalamin receptor defect
Methylmalonate semialdehyde dehydrogenase deficiency
Propionicacidemia
Isovaleric acidemia
Glutaricaciduria, type I
3-Methylcrotonyl-CoA carboxylase 1 deficiency
3-Methylcrotonyl-CoA carboxylase 2 deficiency
3-methylglutaconic aciduria, type I
Barth syndrome
HMG-CoA lyase deficiency
Holocarboxylase synthetase deficiency
Biotinidase deficiency
Beta-ketothiolase deficiency
2-methylbutyrylglycinuria
Isobutyryl-CoA dehydrogenase deficiency
L-2-hydroxyglutaric aciduria
Ethylmalonic encephalopathy
Malonyl-CoA decarboxylase deficiency
CPT II deficiency
Carnitine-acylcarnitine translocase deficiency
Acyl-CoA dehydrogenase, short-chain, deficiency of
Acyl-CoA dehydrogenase, medium chain, deficiency of
VLCAD deficiency
LCHAD deficiency
Trifunctional protein deficiency
Glutaric acidemia II
3-hydroxyacyl-CoA dehydrogenase deficiency
2,4-dienoyl-CoA reductase deficiency
Mucopolysaccharidosis Ih / Ih/s /Is
Mucopolysaccharidosis II
Mucopolysaccharidosis type IIIA (Sanfilippo A)
Mucopolysaccharidosis type IIIB (Sanfilippo B)
Mucopolysaccharidosis IVA
GM1-gangliosidosis, type I / II /III
Mucopolysaccharidosis VII
Mucopolysaccharidosis type VI (Maroteaux-Lamy)
Niemann-Pick disease, type A / B
Niemann-Pick disease, type C1
Niemann-pick disease, type C2
Gaucher disease, perinatal lethal
Fabry disease
Tay-Sachs disease
Krabbe disease
Krabbe disease, atypical
Metachromatic leukodystrophy
Glycogen storage disease II
Mucolipidosis II alpha/beta, III alpha/beta
Glycogen storage disease Ia
Glycogen storage disease Ib / Ic
Glycogen storage disease IIIa / III b
Glycogen storage disease VI
Glycogen storage disease, type IX
Galactosemia
Galactokinase deficiency with cataracts
Galactose epimerase deficiency
Fructose intolerance, hereditary
Pyruvate carboxylase deficiency
Epilepsy, pyridoxine-dependent
Pyridoxamine 5'-phosphate oxidase deficiency
GLUT1 deficiency syndrome
Neu-Laxova syndrome 1
Phosphoserine aminotransferase deficiency
Phosphoserine phosphatase deficiency
Cerebral creatine deficiency syndrome 3
Cerebral creatine deficiency syndrome 2
Cerebral creatine deficiency syndrome 1
Segawa syndrome, recessive
Aromatic L-amino acid decarboxylase deficiency
Adrenal hyperplasia, congenital, due to 11-beta-hydroxylase deficiency
HSD10 mitochondrial disease
Adrenoleukodystrophy
Wilson disease
Menkes disease
Achondroplasia
Hemolytic anemia due to G6PD deficiency
Crigler-Najjar syndrome, [Gilbert syndrome]
Genetic disease
Hemophilia B
B
Deaf
Deaf
Hemophilia B attribute is sex chromosome inheritance, characterized by impairment of activated partial thromboplastin, prolonged coagulation time, bleeding symptoms are mild than Hemophilia A, and spontaneous bleeding can occur in severe patients without obvious trauma.
Genetic diagnosis of hemophilia is an effective and accurate method, and is currently mainly analyzed by PCR.
Deafness genetic testing is through the detection of human DNA, found whether the existence of deafness gene mutation sites. For the family has congenital deafness members or thus clear the cause, has a good preventive significance for the reoccurrence of deafness
Congenital deaf children with genetic factors can be detected early through genetic testing. Early intervention and rehabilitation measures to effectively avoid prelingual deafness, children with potential drug-induced deafness can be found, and give clear and targeted drug guidance and tips to avoid drug-induced deafness.
The Advantage of Product


Profession
A genetic testing package designed for newborn screening

Fast
5 working days to report,Respond quickly to clinical needs

Accuracy
Multiple quality control,Multiple center validation,a leading domestic phenotype-genotype database
Suitable for this test
1.Newborns with abnormal results of routine biochemical screening and tandem mass spectrometry;
2.Newborns with abnormal hearing screening who failed
3.Newborns with clinical manifestations such as delayed jaundice,feeding difficulties,vomiting,diarrhea,anemia,etc;
4.Children in NICU and PICU;
5.All newborns.
Sample requirements

Dry blood spots
(Five 3×2mm blood spots)
Peripheral Blood
(1ml)
The Process of Detection
Consultation and informed consent

Collection of sample
Send of sample

Detection of sample

Send of Report

Consultion of Inheritance


Sample receipt and information input
DNA extraction and library building

Capture the library

High-throughput sequencing

Information analysis

Automated report

Report review
Case sharing


Abnormal results of tandem mass spectrometry for genetic metabolic disease in a 4-day-old baby: Phe and Phe/Tyr are both high, and different pathogenic genes correspond to different treatment plans.

Babies and their parents use “Badyhealth”newborn screening of inherited metabolic diseases by NGS: The pathogenic locus where the baby carries the PAH gene was found.

Baby diagnosis: Phenylalanine Hydroxylation Deficiency Disease.

Timely and effective treatment: The baby has a good prognosis, and the intellectual development is close to normal people.
HPA is classified into two major groups based on etiology: phenylalanine hydroxylase deficiency and PAH coenzyme biotrexate (BH4) deficiency, but the two disorders are treated differently. If not early diagnosis and early treatment, will bring irreversible harm to the baby. Therefore, it is very important to find the cause of the disease timely and accurately by NGS, assist clinicians to diagnose the disease, and make the newborn receive the effective treatment in the first time.
Related products
Newborn Screening of Inherited Metabolic Diseases by NGS




















