





.png)




















2021-12-31
导读
串联质谱(MS/MS)技术已经广泛应用于新生儿遗传代谢病筛查项目,但存在假阳性率较高的问题。根据大样本汇总分析的结果,我国串联质谱技术用于新生儿遗传代谢病筛查的初筛阳性率约为1.91%,而最终确诊阳性率仅为0.04%,即每1万例新生儿中,约有191例初筛为阳性,经召回复查及进一步检测,最终仅有4例确诊。造成串联质谱筛查假阳性结果的因素有很多,本期我们探讨携带者状态对于筛查结果的影响。
什么是携带者状态
绝大多数遗传代谢病为常染色体隐性遗传模式,即只有当同源染色体上的两份基因拷贝均发生变异时才会导致疾病的发生,而仅有1份拷贝发生变异不会致病,此时称之为携带者(carrier)或携带者状态(carrier status)。
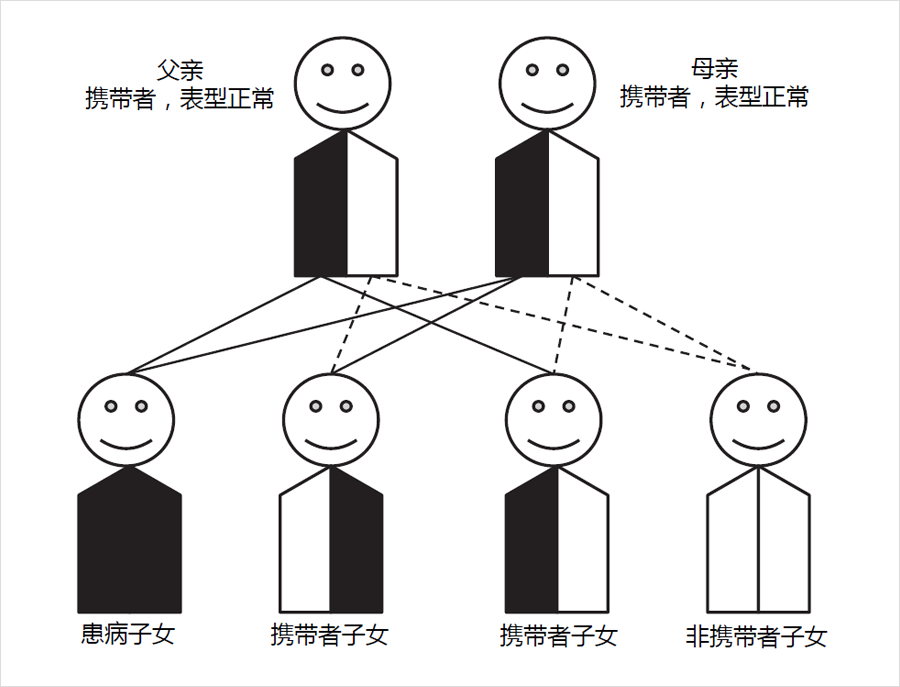
携带者状态对新生儿串联质谱筛查结果的影响
由于正常等位基因的剂量补偿效应,遗传代谢病基因变异携带者不会患病,但与完全正常人群相比,其酶活性或多或少会受到一定程度的影响。对于新生儿遗传代谢病串联质谱筛查,这种影响可能会体现在特定疾病相应指标出现异常,尤其是对于那些指标特异性较差的疾病,如中链酰基辅酶A脱氢酶缺乏症(MCADD)和极长链酰基辅酶A脱氢酶缺乏症(VLCADD)等。
01 中链酰基辅酶A脱氢酶缺乏症(MCADD)
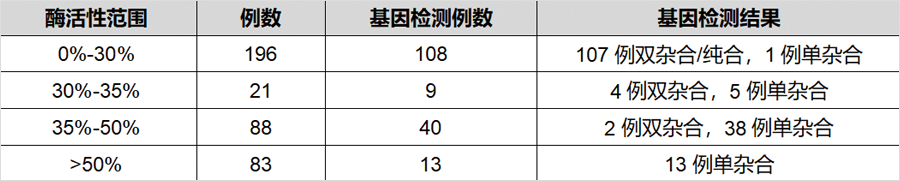
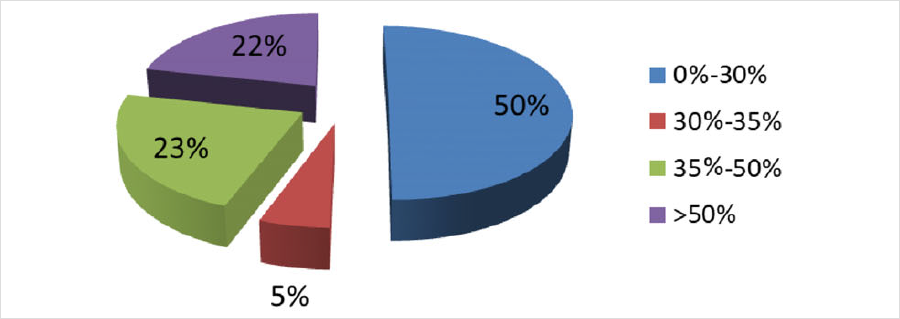
发表在《J Inherit Metab Dis》杂志,题为“Genotype and residual enzyme activity in medium-chain acyl-CoA dehydrogenase (MCAD) deficiency: Are predictions possible?”的研究[1],报道了460例串联质谱筛查疑似MCADD的新生儿,通过酶活性检测和/或基因检测进行确诊,并分析了基因型和酶活性的相关性。在388例进行了酶活性检测的新生儿中,196例酶活性介于0%-30%(疑似患者),21例酶活性介于30%-35%(疑似患者或携带者),88例酶活性介于35%-50%(疑似携带者),其余酶活性>50%(疑似正常)。171例新生儿同时进行了基因检测(44%),确诊57例为携带者。粗略推算因携带者状态导致的假阳性比例在40%以上。
02 极长链酰基辅酶A脱氢酶缺乏症(VLCADD)
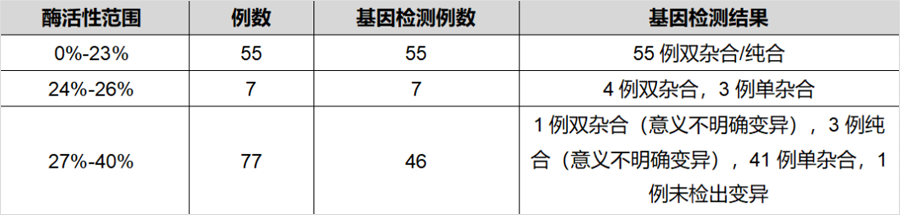
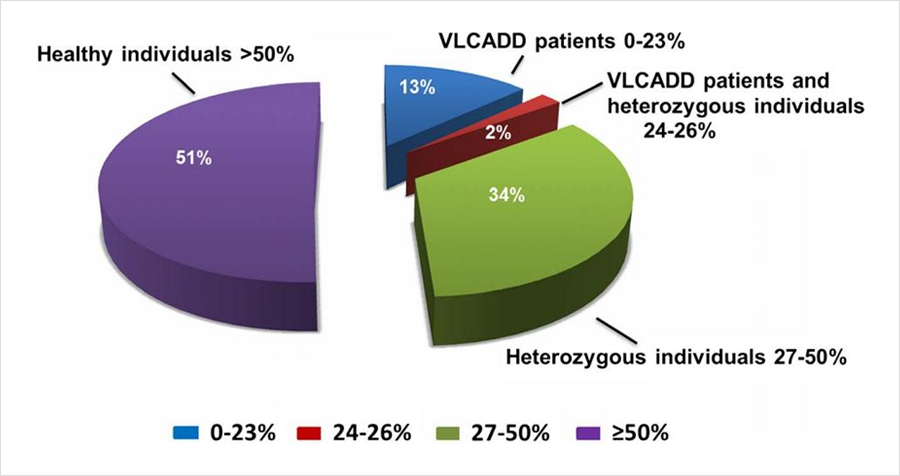
同样发表在《J Inherit Metab Dis》杂志,题为“The diagnostic challenge in very-long chain acyl-CoA dehydrogenase deficiency (VLCADD)”的研究[2],报道了403例串联质谱筛查疑似VLCADD的新生儿,采用酶活性检测和/或基因检测进行确诊。55例酶活性介于0%-23%(疑似患者),7例酶活性介于24%-26%(疑似患者或携带者),138例酶活性介于27%-50%(疑似携带者),203例酶活性>50%(疑似正常)。108例酶活性<40%的新生儿同时进行了基因检测,确诊44例为携带者。粗略推算因携带者状态导致的假阳性比例接近40%。
03 血酰基肉碱水平是否可提示携带者状态?
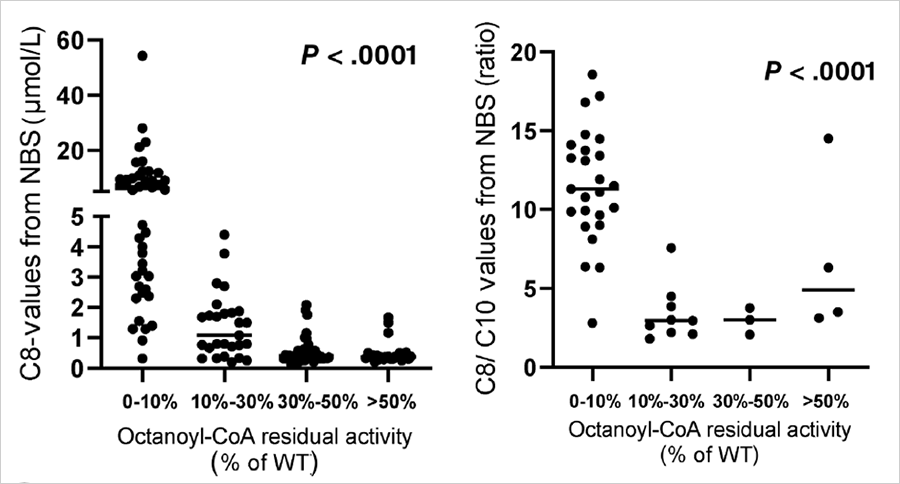
血辛酰肉碱(C8)及C8/C10比值增高是串联质谱筛查MCADD的主要诊断指标,那这两个指标是否可用来初步区分患者与携带者呢?在上述的MCADD研究中,研究人员做了酶活性与各个指标相关性的分析。如下图所示,酶活性介于0%-10%的MCADD患者,其串联质谱C8和C8/C10值均显著高于其他个体,但存在一定程度的交叉;而酶活性介于10%-30%的患者,与携带者和正常个体相比,没有显著差异。因此,通过C8和C8/C10数值进行患者和携带者的区分是不可行的。
思考与总结
携带者状态是导致新生儿串联质谱MCADD和VLCADD筛查假阳性结果的主要原因之一。由于酶活性检测的普及率较低,基因检测是目前确诊的主要手段。但汇总既往研究报道[3-5],我国MCADD患儿基因诊断的灵敏度仅为78.9%(汇总分析的38例经临床、血生化、血肉碱或尿有机酸检测等确诊的患儿中,8例仅检测到单杂合变异),这与本文提到的国外研究存在较大差异。因此,在应用基因检测技术对串联质谱筛查阳性的MCADD患儿进行辅助诊断时,如果仅检出单杂合变异,除考虑携带者状态造成的假阳性可能外,也不能排除基因检测假阴性的可能。而对于VLCADD,我国患儿基因诊断的灵敏度接近100%[6-8],因此,检出单杂合变异时不需要考虑假阴性的可能。
参考文献:
1. Tucci S, Wagner C, Grünert SC, et al. Genotype and residual enzyme activity in medium-chain acyl-CoA dehydrogenase (MCAD) deficiency: Are predictions possible?. J Inherit Metab Dis. 2021;44(4):916-925.
2. Hesse J, Braun C, Behringer S, Matysiak U, Spiekerkoetter U, Tucci S. The diagnostic challenge in very-long chain acyl-CoA dehydrogenase deficiency (VLCADD). J Inherit Metab Dis. 2018;41(6):1169-1178.
3. Gong Z, Liang L, Qiu W, et al. Clinical, Biochemical, and Molecular Analyses of Medium-Chain Acyl-CoA Dehydrogenase Deficiency in Chinese Patients. Front Genet. 2021;12:577046.
4. Li Y, Zhu R, Liu Y, Song J, Xu J, Yang Y. Medium-chain acyl-coenzyme A dehydrogenase deficiency: Six cases in the Chinese population. Pediatr Int. 2019;61(6):551-557.
5. 童凡, 蒋萍萍, 杨茹莱,等. 中链酰基辅酶A脱氢酶缺乏症新生儿筛查及随访研究[J]. 中国当代儿科杂志, 2019, 21(1):6.
6. 曹金俊, 邱文娟, 章瑞南,等. 极长链酰基辅酶A脱氢酶缺乏症11例的临床和ACADVL基因突变谱分析[J]. 中华儿科杂志, 2015(4):6.
7. Li X, Ma R, Liu Y, et al. One potential hotspot ACADVL mutation in Chinese patients with very-long-chain acyl-coenzyme A dehydrogenase deficiency. Clin Chim Acta. 2020;503:218-222.
8. 钱古柃,洪芳,童凡,等. 极长链酰基辅酶A脱氢酶缺乏症16例临床及基因型分析[J]. 儿科药学杂志, 2020, 26(9):4.































