.png)













2021-08-11
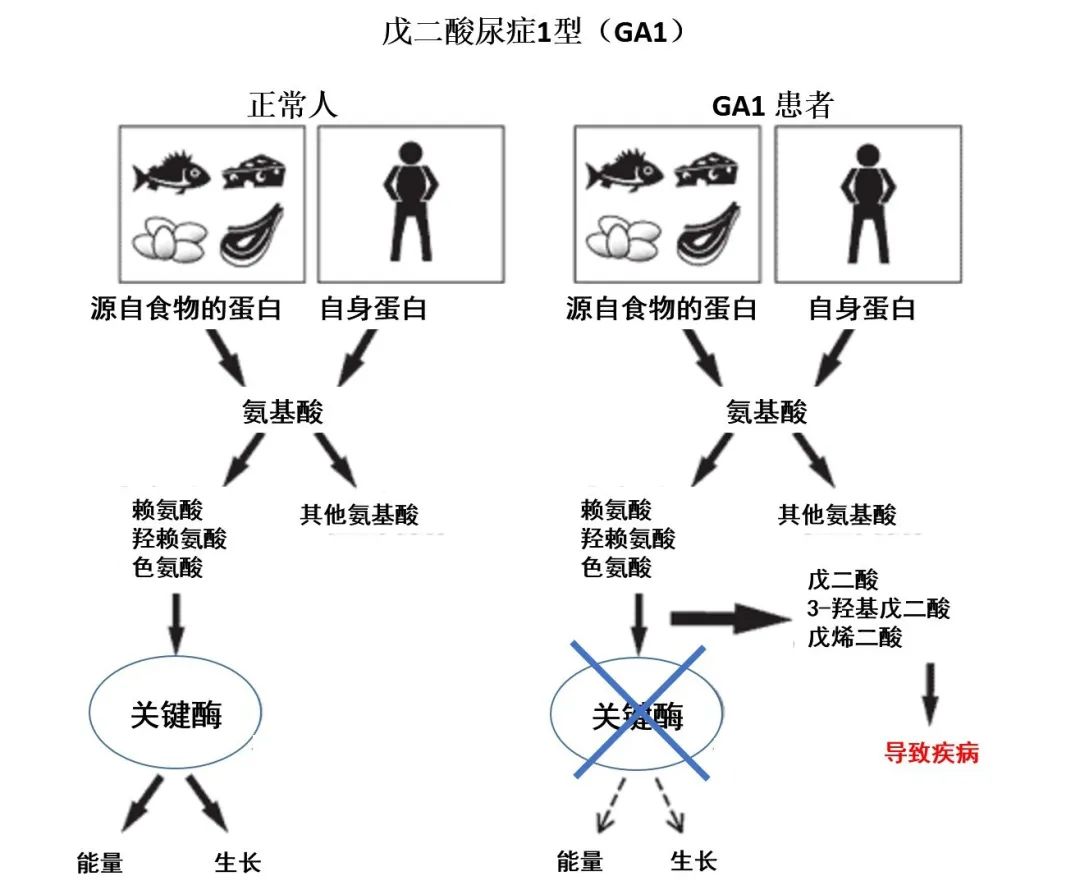
戊二酸血症1型(GA1)是一种常染色体隐性遗传的代谢性疾病,又称为戊二酸尿症1型,是由于戊二酰辅酶A脱氢酶(GCDH)活性降低或缺失,导致赖氨酸、羟赖氨酸及色氨酸分解代谢受阻,代谢产物戊二酰肉碱(C5DC)、戊二酸及3-羟基戊二酸等在体内异常蓄积,引起代谢紊乱。
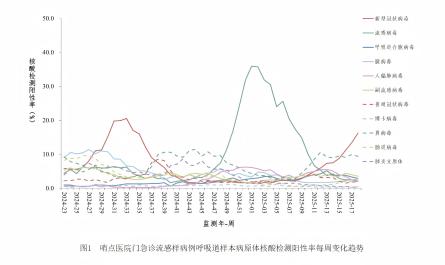
根据大样本新生儿串联质谱遗传代谢病筛查数据,我国GA1的发病率约为1/310 200~1/52078。GA1患者常于婴幼儿期发病,临床表现多样,差异较大,以神经系统表现为主,可伴有其他系统异常。约80-90%的未经治疗患儿将出现神经系统受累表现,常由感染、发热、疫苗接种及手术等诱发急性脑病发作。少部分患者隐匿起病,甚至成年发病,属于晚发型,多表现为非特异性神经系统症状,如头痛、眩晕、共济失调或运动后昏厥、大小便失禁、注意力涣散和感觉异常等,脑白质营养不良多见。
串联质谱筛查的局限性
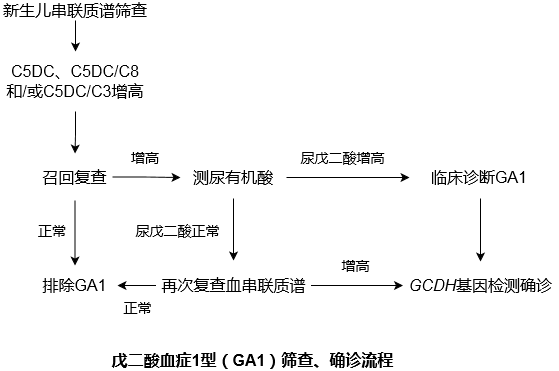
GA1的治疗关键是早诊断和早治疗,如果能在症状出现之前筛查出患者,并及时治疗,可以减轻甚至避免神经系统损害。新生儿遗传代谢病筛查是GA1获得早期诊断的重要前提,串联质谱分析血酰基肉碱水平是目前初筛的首选方法(C5DC、C5DC/C8和/或C5DC/C3增高),尿有机酸分析(戊二酸增高,伴或不伴有3-羟基戊二酸增高)可用于辅助临床诊断,但串联质谱的检测仍然可能造成假阴性与假阳性的情况,所以确诊仍需依赖于GCDH基因检测。
假阴性
根据尿戊二酸水平可将GA1分为低分泌型(<100μmol/mol肌酐)和高分泌型(>100μmol/mol肌酐),对应的残余酶活性分别3~30%和0-2%。尽管低分泌型患者同样易发生神经系统损伤,不能等同于轻症患者,但却是造成串联质谱筛查漏检的主要原因。
根据德国一项多中心队列研究结果,在1999-2016年确诊的94例GA1患者中,67例为高分泌型(71.3%),25例为低分泌型(26.6%),2例未知;87例为新筛发现,4例新筛漏检,3例为新筛发现的母源性GA1患者。由此获得的串联质谱筛查GA1的总体灵敏度为95.6%(87/91),但对于低分泌型患者,灵敏度仅有84%(21/25)。
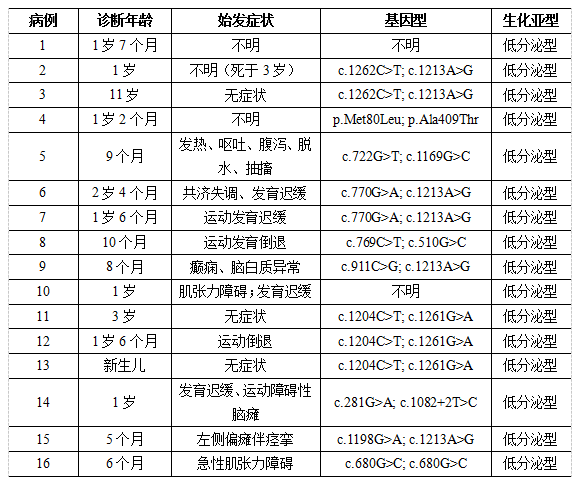
另外2篇发表于2021年2月份和6月份的研究分别报道了5例和4例新筛漏检的GA1患者,均为低分泌型。这些患者及其他个案报道病例的临床信息及基因型信息如下表所示(病例2和3、病例6和7、病例11、12和13为同胞(病例12为先证者))。所有漏检病例均为低分泌型,多数患者表现出GA1的典型症状,少数隐匿发病或至随访结束时仍无症状。
假阳性
串联质谱用于GA1筛查产生假阳性的常见原因:
(1)母源性GA1;
(2)新生儿肾功能障碍;
(3)戊二酸血症2型(多种酰基辅酶A脱氢酶缺乏症);
(4)中链酰基辅酶A患儿的假性戊二酰血症等。
GA1的基因筛查
GA1致病基因为GCDH,目前全世界已报道的致病变异超过200多种。我们搜集整理了文献报道的84例中国GA1患者的基因检测结果,并进一步获得了GCDH基因变异谱数据,如下表所示。84例患者均检测到GCDH基因双杂合或纯合变异,基因诊断率为100%;共检出61种基因变异类型,主要为点突变,少数为小片段插入/缺失变异;c.1244-2A>C变异占比最高(25.5%),且主要见于南方地区的患者;多数占比较高的变异与高分泌型GA1相关,但c.892G>A和c.482G>A变异与低分泌型相关。
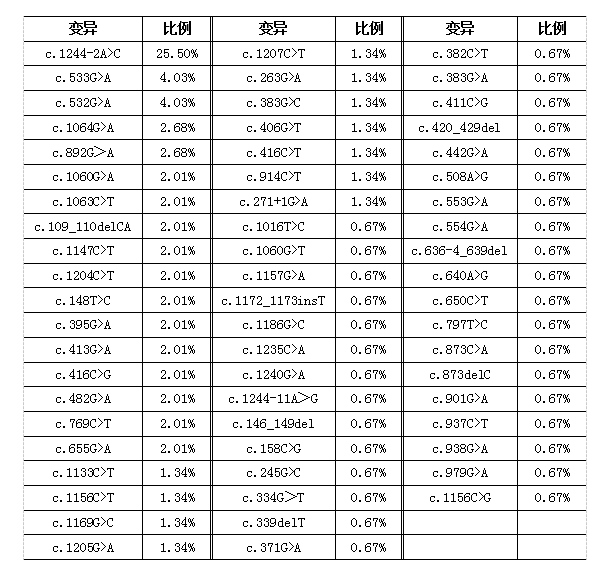
基于GCDH基因变异特征,理论上通过基因检测进行新生儿GA1筛查的灵敏度可接近100%,优于串联质谱筛查。早在2002年,即有特定人群GA1基因筛查研究的报道。随着基因测序技术的快速发展、医学遗传学研究的不断深入,包含GA1在内的遗传代谢病高通量基因筛查研究在国内外广泛开展,其可作为当前一级筛查计划的补充,提高筛查的灵敏度和特异度,简化疾病确诊流程,缩短确诊时间。我们已有的筛查数据显示,GCDH基因c.892G>A(低分泌型)变异在普通人群中的携带率仅次于c.1244-2A>C变异,居第二位,也是排在第三位的c.532G>A变异的2倍。这与临床确诊病例的基因变异谱之间存在差异,可能与标本来源偏倚有关,或提示c.892G>A变异相关的GA1患者在新生儿串联质谱筛查或临床诊疗中存在漏检可能。
联合医学新生儿遗传和代谢病基因检测
联合医学“贝可康”新生儿遗传和代谢病基因检测产品,采用“All-In-One”超高重PCR扩增子捕获的专 利技术和二代高通量测序技术,检测新生儿130个与遗传代谢病相关的基因,对新生儿可干预的126种遗传和代谢病进行检测,早发现、早诊断、 早治疗,降低疾病对宝宝的伤害。